Research
Genetic associations with meiotic aneuploidy in day-5 embryos
Aneuploidy (gain or loss of whole chromosomes) is the leading cause of pregnancy loss and congenital disorders. Only approximately half of all conceptions survive to live birth, primarily due to aneuploidy.
We conduct a retrospective analysis of preimplantation genetic testing for aneuploidy (PGT-A) data from 139,416 in vitro fertilized embryos (22,850 sets of biological parents), identifying 3,656,198 meiotic crossovers and 92,485 aneuploid chromosomes. We find that aneuploid embryos are depleted of crossovers relative to euploid embryos.
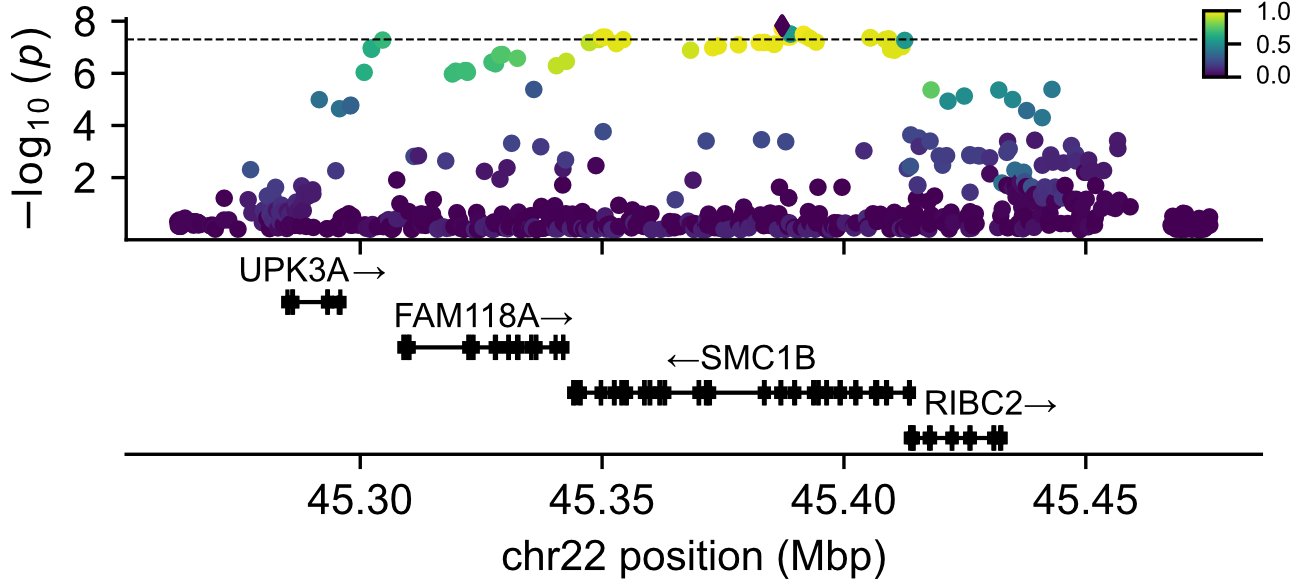
I identify a genome-wide association with a haplotype spanning several genes, including SMC1B, which encodes a meiotic cohesin protein. Transcriptome-wide association (comparing gene expression data from GTEx to our detected rates of aneuploidy) recapitulated this association, as well as two others, identifying C14orf39 (which encodes a unit of the synaptonemal complex) and NCAPD2 (which encodes a regulatory subunit of the condensin I complex) as genes whose expression levels associate with aneuploidy risk.
This work was accepted to Nature (with an expected publication date of January 2026) and is available as a preprint in medRxiv (Carioscia et al. 2025), where we explore further detail about a putative regulatory mechanism by which this haplotype modulates aneuploidy.
Estimating rates of meiotic and mitotic error in human development
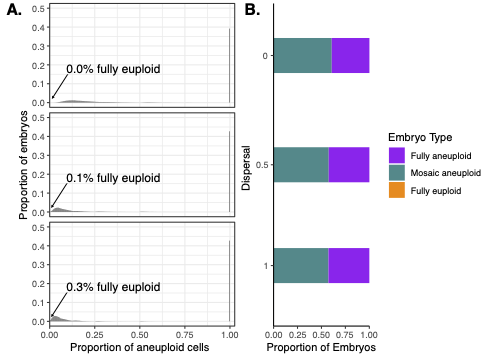
Estimates of euploid, aneuploid, and mosaic (embryos comprising a mix of euploid and aneuploid cells) of embryos are based on clinical classifications of PGT-A. However, PGT-A relies on a biopsy of just a few cells from the multi-cell day-5 embryo, which may not accurately reflect the true nature of each embryo.
As a result, these data also do not offer adequate foundation to estimate the rates of meiotic and mitotic error actually occurring in human development. Here, we conduct simulations across a range of each error rate, simulate biopsies, and use approximate Bayesian computation to identify the set of simulated embryos that most matches clinical data. The results offer us ranges of these error rates that best explain the clinical data observed through PGT-A.
This project began during the rotation of Matthew Isada, a PhD student in the Biology Department at JHU who I mentored in 2022. I then mentored undergraduate Angela Yang as she scaled up our initial 2D and 3D simulations of embryo development and biopsy to investigate a range of errors across numerous replicates.
This work is available in _Genetics_ (Yang, Carioscia et al. 2025), where we estimate the true incidence of mosaicism at the day-5 stage and investigate a range of misclassification parameters to offer a robust analysis of clinical data.
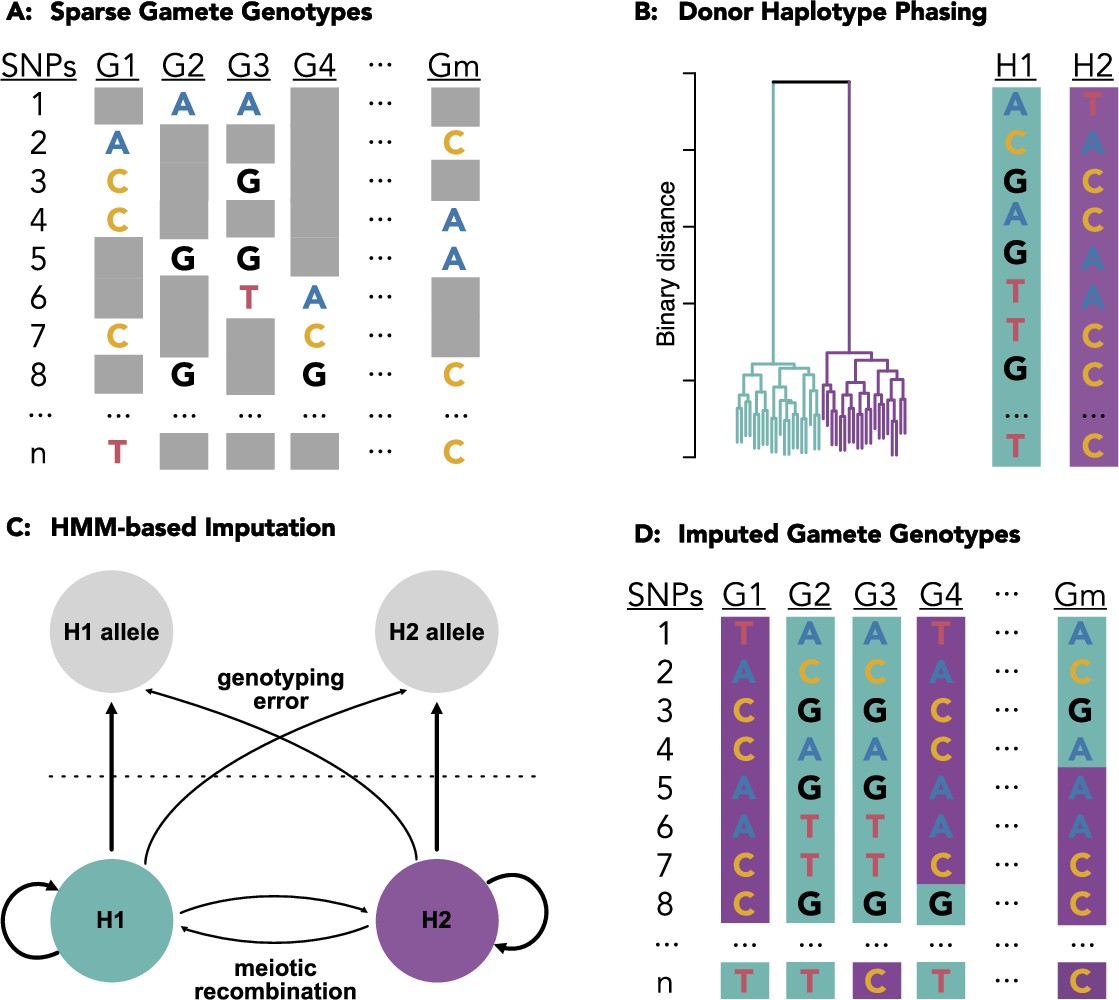
rhapsodi: an R package to impute sparsely sequenced haploid genomes
The recent development of high-throughput single-cell genome sequencing of human sperm (termed "Sperm-seq") offers an opportunity to study various aspects of meiosis and inheritance with improved statistical power. However, the low sequencing coverage per cell (0.01x) necessitates the development of tailored statistical methods for recovering gamete genotypes.
To this end, we developed a method called rhapsodi (R haploid sperm/oocyte data imputation) that uses low-coverage single-cell DNA sequencing data from large samples of gametes to reconstruct phased donor haplotypes, impute gamete genotypes, and map meiotic recombination events.
We then applied rhapsodi to investigate evidence of biases in human inheritance. Mendel’s Law of Segregation states that the offspring of a diploid, heterozygous parent will inherit either allele with equal probability. While the vast majority of loci adhere to this rule, research in model and non-model organisms has uncovered numerous exceptions whereby “selfish” alleles are disproportionately transmitted to the next generation. Evidence of such transmission distortion (TD) in humans remains equivocal in part due to statistical and sample size limitations of past studies.
We leveraged single-cell sequencing data from over 40,000 individual sperm across 25 donors (5 donors with known infertility phenotypes and 20 donors of presumed normal fertility). This scan enabled us to look at an earlier developmental time point than had been considered in previous studies, with the possibility of avoiding the effects of downstream selection and other biases. Our results exhibited close concordance with binomial expectations under balanced transmission, in contrast to tenuous signals of TD that were previously reported in pedigree-based studies.
Our paper introduces and benchmarks rhapsodi and then applies it to this data in investigation of transmission distortion (Carioscia et al. 2022). This work was highlighted in an eLife digest! See our paper and our code!